
Samples quality control tests
User can add samples tests to be performed in this steps. The order of the parameters in the Aozan configuration file will be reused in the report generated by this step. Each test can use several parameters and contains always an "enable" property to activate the test. Usually a property named "interval" exists for each test, however if this property is not set, the value will be computed but not checked if it is the correct interval. You will find below all the parameters for all the available tests in Aozan.
For the quality control sample test, the report brings together three source of data :
- Data supplied by the demultiplexing step
- FastQC
- Contamination detection with FastQ Screen
- Analysis undetermined clusters from demultiplexing.
Sample quality tests from demultiplexing step
| Aozan property | Type | Default value | description |
|---|---|---|---|
| qc.test.sample.genome.names.enable | boolean | False | Enable genome names test |
| qc.test.sample.raw.cluster.count.enable | boolean | False | Enable raw clusters count test |
| qc.test.sample.raw.cluster.count.interval | integer interval | False | Interval for valid values |
| qc.test.sample.pf.cluster.count.enable | boolean | False | Enable passing filter count test |
| qc.test.sample.pf.cluster.count.interval | integer interval | False | Interval for valid values |
| qc.test.sample.pf.percent.enable | boolean | False | Enable passing filter percent test |
| qc.test.sample.pf.percent.interval | double interval | False | Interval for valid values |
| qc.test.sample.in.lane.percent.enable | boolean | False | Enable in lane percent test |
| qc.test.sample.in.lane.percent.distance | double | 0.05 | Interval for valid values of in lane percent tet. The interval is set according to the theoretical percent (1.0/sampleCount) |
| qc.test.sample.q30.percent.enable | boolean | False | Enable Q30 percent test |
| qc.test.sample.q30.percent.interval | double interval | False | Interval for valid values |
| qc.test.sample.base.pf.mean.quality.score.enable | boolean | False | Enable base passing filter mean quality sample test |
| qc.test.sample.base.pf.mean.quality.score.interval | double interval | False | Interval for valid values |
| qc.test.sample.recoverable.raw.cluster.count.enable | boolean | False | Enable recoverable raw cluster count sample test: compute raw cluster count that can be recovered from the undetermined FASTQ file with more one mismatch that has been used in the demultiplexing step. |
| qc.test.sample.recoverable.raw.cluster.count.interval | integer interval | False | Interval for valid values |
| qc.test.sample.recoverable.pf.cluster.count.enable | boolean | False | Enable recoverable passing filter cluster sample test: compute passing filter cluster count that can be recovered from undetermined FASTQ file with more one mismatch that has been used in the demultiplexing step. |
| qc.test.sample.recoverable.pf.cluster.count.interval | integer interval | False | Interval for valid values |
| qc.test.sample.cluster.recovery.report.enable | boolean | False | Enable recovery report |
The explanation to construct intervals is here.
Sample quality tests from FastQC
Aozan launch FastQC on each sample (using parallel computation) and creates a quality control report file. If no FastQC test property is enabled, FastQC will not be launched on FASTQ files.
Configuration tests
| Aozan property | Type | Default value | description |
|---|---|---|---|
| qc.test.sample.fastqc.basic.stats.enable | boolean | False | Enable FastQC basic stats test |
| qc.test.sample.fastqc.per.base.quality.scores.enable | boolean | False | Enable FastQC per base quality scores test |
| qc.test.sample.fastqc.per.sequence.quality.scores.enable | boolean | False | Enable FastQC per sequence quality scores test |
| qc.test.sample.fastqc.per.base.sequence.content.enable | boolean | False | Enable FastQC per base sequence content test |
| qc.test.sample.fastqc.per.sequence.gc.content.enable | boolean | False | Enable FastQC per sequence GC ccontent test |
| qc.test.sample.fastqc.n.content.enable | boolean | False | Enable FastQC ncontent test |
| qc.test.sample.fastqc.sequence.length.distribution.enable | boolean | False | Enable FastQC sequence length distribution test |
| qc.test.sample.fastqc.duplication.level.enable | boolean | False | Enable FastQC duplication level test |
| qc.test.sample.fastqc.overrepresented.sequences.enable | boolean | False | Enable FastQC overrepresented sequences test |
| qc.test.sample.fastqc.kmer.content.enable | boolean | False | Enable FastQC kmer content test |
| qc.test.sample.fastqc.per.tile.sequence.quality.enable | boolean | False | Enable FastQC per tile sequence quality test |
| qc.test.sample.fastqc.adapter.content.enable | boolean | False | Enable FastQC adapter content test |
General configuration of FastQC
| Aozan property | Type | Default value | description |
|---|---|---|---|
| qc.conf.fastqc.contaminant.file | string | Not set | Specifies a file path which contains the list of contaminants to screen overrepresented sequences. The content of the file will override the default list of contaminants. The file must contain sets of named contaminants in the form name[tab]sequence. Lines prefixed with a hash will be ignored. (FastQC -c / --contaminants parameter) |
| qc.conf.fastqc.kmer.size | integer | 7 | Specifies the length of Kmer to look for in the Kmer content module. Specified Kmer length must be between 2 and 10. (FastQC -k / --kmers parameter) |
| qc.conf.fastqc.nogroup | boolean | False | Disable grouping of bases for reads >50bp. (FastQC --nogroup parameter) |
| qc.conf.fastqc.adapter.file | string | No set | Path to a specific adapter file, replace the default adapter file. (FastQC -a / --adapters parameter) |
| qc.conf.fastqc.limits.file | string | No set | Path to a specific limits file, replace the default file. (FastQC -l / --limits parameter) |
| qc.conf.fastqc.expgroup | boolean | False | Use exponential base groups in graph. (FastQC parameter) |
| qc.conf.fastqc.casava | boolean | False | Illumina FASTQ file. (FastQC --casava parameter) |
| qc.conf.fastqc.nofilter | boolean | False | Option for filtering bad Illumina FASTQ entries. (FastQC parameter) |
| qc.conf.fastqc.process.undetermined.samples | boolean | True | Run FastQC on undetermined indices samples, default at false. |
| qc.conf.fastqc.unzip.report.file | boolean | False | Unzip the FastQC Zip report file. |
| qc.conf.fastqc.keep.zip.report.file | boolean | True | Keep the FastQC Zip report file. |
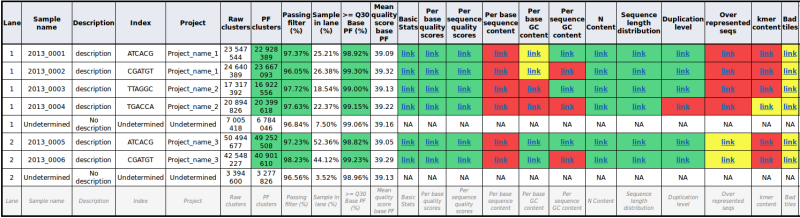
An example of table built with sample quality tests
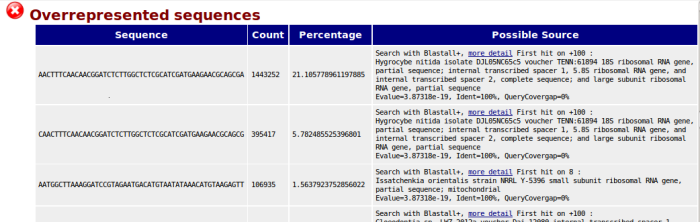
Enhancement to module ‘Overrepresented sequences’
The output of ‘Overrepresented Sequences’ module from FastQC has been improved for sequences not found in contaminant list. We (optionally) launch a blast on sequences with ‘no hit’ source. The report prints the best hit.
The blast execution can be disabled if an Aozan section with a disable.blast is set to true in the samplesheet of the run.
| Aozan property | Type | Default value | description |
|---|---|---|---|
| qc.conf.fastqc.blast.enable | boolean | false | Enable the blast search if not hit found for an overrepresented sequence |
| qc.conf.fastqc.blast.path | string | /usr/bin/blastn | Path of the blast executable. It can run blastn from ncbi-blast+ or blastall from blast2 |
| qc.conf.fastqc.blast.db.path | string | /home/aozan/ncbi_database_nt/nt | Path to the nt database (where is located the nt.nal file) |
| qc.conf.fastqc.blast.arguments | string | Not set | Blast parameters. DO NOT use the following blast options: -d (blastn), -db (path to database), -outfmt (output type, must be in xml) and -num-threads (number processor) |
A Docker container can be use to launch Blast. When the Docker is enabled for Blast, Blast2 2.2.26 will be use.
| Aozan property | Type | Default value | description |
|---|---|---|---|
| qc.conf.fastqc.blast.use.docker | boolean | false | Use Docker container to launch Blast |
| docker.uri | string | unix:///var/run/docker.sock | Docker deamon URI |
Example of new module ‘Overrepresented sequences’:
Sample quality tests from contamination detection
Aozan includes a fast Java FastQ Screen implementation. This module maps sample reads on a list of reference genomes for assessing sample contamination and the ratio of the excepted genome in the sample. It creates a report file with values for each genome.
This step assesses the sample contamination from the FASTQ files. To do this, Aozan uses the FastQ Screen strategy. FastQ Screen allows to screen a library of sequences in Fastq format against a set of sequence databases.
The FastQ Screen code has been rewritten for Aozan and additional values are now calculated. Here we use bowtie to align reads on genomes or on contaminant databases like adapter sequences, PhiX and ribosomes databases. Moreover it aligns reads samples on the genome defined in the bcl2fastq sample sheet file.
To avoid too long computation, the mapping on genomes is limited to a small set of the reads of the FASTQ files. By default 200,000 PF reads (passing illumina filters) are used for each sample (It is enough for estimate the contamination in a sample).
Aozan uses Eoulsan to handle alignments and especially the repository system for genomes and genomes indexes.
If no FastQ Screen test property is enabled, FastQ Screen will not be launched on FASTQ files.
Configuration tests
| Aozan property | Type | Default value | Description |
|---|---|---|---|
| qc.test.sample.fastqscreen.mapped.except.ref.genome.percent.enable | boolean | False | Enable the percent of reads mapped at least one reference genome, except on genome sample test |
| qc.test.sample.fastqscreen.mapped.except.ref.genome.percent.interval | double interval | [0, 0.1] | Interval of valid values |
| qc.test.sample.fastqscreen.mapped.percent.enable | boolean | False | Add columns in the report with the percent of reads mapped for each reference genome |
| qc.test.sample.fastqscreen.mapped.percent.interval | double interval | [0, 0.1] | Interval of valid values |
| qc.test.sample.fastqscreen.report.enable | boolean | False | Enable FastQ Screen full report link column |
The explanation to construct intervals is here.
General configuration for launch the mapper
| Aozan property | Type | Default value | Description |
|---|---|---|---|
| qc.conf.fastqscreen.genome.aliases.path | string | Not set | Path to the file which make the match between genome name in bcl2fastq samplesheet file and the genome name used for bowtie index. If a genome name does not exist in the alias file, it will be added at the end of the file |
| qc.conf.fastqscreen.genome.descs.path | string | Not set | Path to the genome descriptions repository. The genome description file contains some basic informations about the genome like the names of the chromosome and their lengths. The genome description files allow to avoid useless genome sequence parsing once it has been already parsed forn a previous run |
| qc.conf.fastqscreen.genomes.path | string | Not set | Path to the genomes repository |
| qc.conf.fastqscreen.mapper.indexes.path | string | Not set | Path to the genome indexes repository |
| qc.conf.fastqscreen.genomes | string | phix, adapters | List of reference genomes to always use by fastqscreen |
| qc.conf.fastqscreen.mapper | string | bowtie | In a next version, it will be possible to choice between bowtie and bowtie2. Per default, bowtie is used. |
| qc.conf.fastqscreen.mapper.arguments | string | -l 20 -k 2 --chunkmbs 512 | Arguments of the mapper, in paired-end mode, ‘--maxins 1000’ is added |
| qc.conf.fastqscreen.mapping.skip.control.lane | boolean | True | Enable contamination detection on control lane |
| qc.conf.fastqscreen.mapping.ignore.paired.end.mode | boolean | True | If true for a run paired-end, the detection contamination will be only performed on the first of the two reads. The values for the second read will be the same as first read |
| qc.conf.fastqscreen.fastq.max.reads.parsed | integer | 200000 | Number of reads to use for each mapping. Only the reads with Illumina passing filter will be selected. The selected reads will be written in a dedicated temporary file. If value is set to -1, all reads of the FASTQ files will be used |
| qc.conf.fastqscreen.fastq.reads.pf.used | integer | 30000000 | The temporary FASTQ files are created by parsing at most this maximum number of reads in the FASTQ file source. If value is set to -1, it browses all the entries of the FASTQ files |
| qc.conf.fastqscreen.max.read.length | integer | -1 | The maximun read length to use with FastQ Screen. When enabled, this option will trim the end of the reads if their length is greater than the setting. If value is set to -1, full read length will be used |
| qc.conf.fastqscreen.xsl.file | string | Not set | Path to a specific XSL stylesheet file to use for creating the FastQ Screen HTML report |
| qc.conf.fastqscreen.process.undetermined.samples | boolean | false | Run FastQ Screen on undetermined indices FASTQ files on all genomes available for the run |
An example of a HTML report of quality control with major values of FastQ Screen:
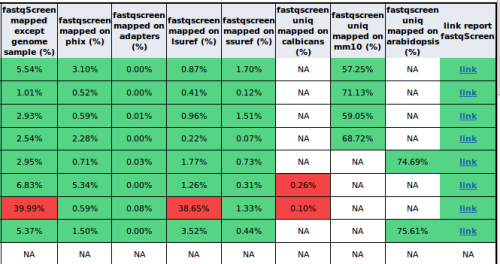
When the use click on a link of the last column, it will be redirected to a dedicated FastQ Screen HTML report. The following figure show a HTML FastQ Screen report with four reference genomes and the genome sample (mm10):
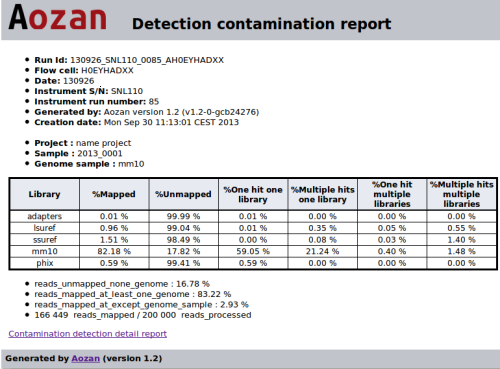
This report contains for each genome the following values:
- %Mapped: the percent of reads which are mapped on this genome;
- %Unmapped: the percent of reads which are not mapped on this genome;
- %One_hit_one_library: the percent of reads which are mapped with one hit on one genome;
- %Multiple_hits_one_library: the percent of reads which are multi-mapped on this genome;
- %One_hit_multiple_libraries: the percent of reads which are mapped with one hit on several genomes;
- %Multiple_hits_multiple_libraries: the percent of reads which are multi-mapped on several genomes.
NB: The genome of the sample is used as a positive control.
List of values for all genomes :
- % reads_unmapped_none_genome: the percent of reads which are not mapped on any genome tested;
- % reads_mapped_at_least_one_genome: the percent of reads which are mapped on at least one genome;
- % reads_mapped_except_genome_sample: the percent of reads which are mapped on the contamination genomes, the genome of the sample is excluded;
Analysis of the undetermined clusters from demultiplexing.
The undetermined FASTQ files created by the demultiplexing step can be analyzed using 3 modules:
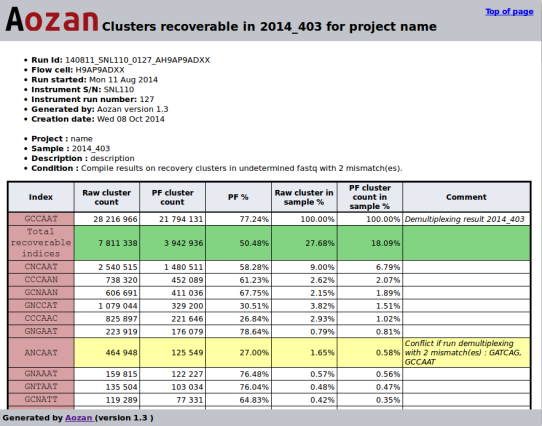
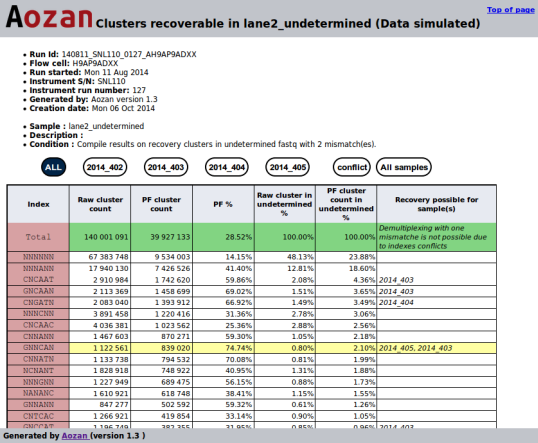
- The recoverable sample clusters module can suggest for each indices, the sample(s) and the number of clusters that can be recovered using a demultiplexing step with one more mismatch that the number of mismatches has been used in the demultiplexing step. This module create two files (one for the lanes, the other for the samples) that display for each index the potential sample(s) and the number of recoverable clusters found.
- The FastQC module. Use the "
qc.conf.fastqc.process.undetermined.samples" property to enable this module. - The FastqScreen module. Use the "
qc.conf.fastqscreen.process.undetermined.samples" property to enable this module.
| Aozan property | Type | Default value | description |
|---|---|---|---|
| qc.test.sample.recoverable.pf.cluster.count.enable | boolean | False | Compute the passing filter clusters that can be recovered from the undetermined FASTQ files with one more mismatch that the number of mismatches used in demultiplexing step (the maximum number of mismatches is 2) |
| qc.test.sample.recoverable.pf.cluster.count.interval | interval | False | Interval for valid values |
| qc.test.sample.cluster.recovery.report.enable | boolean | False | Link to the HTML recoverable clusters report |
The two reports are generated in CSV and HTML formats:
- Example of a recoverable clusters lanes report:
- Example of a recoverable clusters sample report:
Syntax of the interval values
User can define intervals as the next examples:
- [,10] From - infinite to 10 (included)
- [,10[ From - infinite to 10 (excluded)
- [,10) From - infinite to 10 (excluded)
- ]2,10[ From 2 (excluded) to 10 (excluded)
- (2,10[ From 2 (excluded) to 10 (excluded)
- [2,10] From 2 (included) to 10 (included)
- [2, ] From 2 (included) to + infinite